附件1
海藻酸钙等10种食品添加剂新品种
- 一、
-
海藻酸钙(又名褐藻酸钙)
英文名称:Calcium alginate
功能分类:增稠剂、稳定和凝固剂
- (一)
-
用量及使用范围
|
食品分类号 |
食品名称 |
最大使用量/(g/kg) |
备注 |
|
06.03.02 |
小麦粉制品 |
5.0 |
|
|
07.01 |
面包 |
5.0 |
|
(二)质量规格要求
1范围
本质量规格要求适用于从海带(Laminaria)、巨藻(Macrocystis)、泡叶藻(Ascophyllum)等褐藻类植物中经提取加工制成的食品添加剂海藻酸钙(又名褐藻酸钙)。
2 分子式
[(C6H7O6)2Ca]n
3 技术要求
3.1感官要求:应符合表1 的规定。
表1 感官要求
|
项 目 |
要 求 |
检验方法 |
|
色泽 |
白色至黄色 |
将适量样品均匀置于清洁、干燥的白瓷盘内,于光线充足、无异味的环境中,观察其色泽和状态。 |
|
状态 |
纤维状或粒状粉末 |
3.2理化指标:应符合表2的规定。
表2 理化指标
|
项 目 |
指标 |
检验方法 |
|
海藻酸钙含量(以氧化钙计,以干基计),w/% |
8.0~13.0 |
附录A中 A.3 |
|
干燥减量,w/% ≤ |
15.0 |
GB 5009.3直接干燥法 |
|
灰分(以干基计),w/% |
10.0~20.0 |
GB 5009.4 a |
|
铅(Pb)/(mg/kg) ≤ |
4.0 |
GB 5009.12 |
|
砷(以As计)/(mg/kg) ≤ |
2.0 |
GB 5009.11 |
|
a灼烧温度为700℃~800℃ |
||
附 录 A
检验方法
A.1 一般规定
本质量规格要求除另有规定外,所用试剂的纯度应在分析纯以上,所用标准滴定溶液、杂质测定用标准溶液、制剂及制品,应按GB/T 601、GB/T 602、GB/T 603的规定制备,试验用水应符合GB/T 6682中三级水的规定。试验中所用溶液在未注明用何种溶剂配制时,均指水溶液。
A.2 鉴别试验
A.2.1 试剂和材料
A.2.1.1 1,3-二羟基萘乙醇溶液(10 g/L):称取约1 g1,3-二羟基萘溶于100 mL无水乙醇,混匀(现用现配)。
A.2.1.2 盐酸。
A.2.1.3 异丙醚。
A.2.2 鉴别
A.2.2.1 可溶性试验
本品不溶于水,但是溶于碱性溶液或者溶于有与钙结合的物质的溶液中,不溶于有机溶剂。
A.2.2.2 海藻酸盐鉴定
取样品5mg放入试管中,加入5mL水,加入1 mL新制的1,3‐二羟基萘乙醇溶液和5 mL浓盐酸摇匀。把上述混合物加热至沸,轻轻煮沸3min,冷却到15℃左右,转移至30 mL的分液漏斗中,容器用5 mL水洗涤,洗液并入分液漏斗中。加入15 mL异丙醚,振摇提取,分取醚层,同时做空白对照,样品管的异丙醚层与对照管比较,应显深紫色。
A.3海藻酸钙含量的测定
A.3.1方法提要
将海藻酸钙试样灰化后,与酸反应形成可溶性的钙盐,利用乙二胺四乙酸二钠标准滴定溶液滴定反应产生的钙离子,折算成海藻酸钙(以氧化钙计,以干基计)的含量。
A.3.2试剂和材料
A.3.2.1 氢氧化钾溶液(2mol/L):精确称取112g氢氧化钾置于1000mL水中,溶解并混匀。
A.3.2.2 混合酸消化液:硝酸、高氯酸以4:1的质量比混合均匀,备用。
A.3.2.3 三乙醇胺溶液(10%):量取10mL三乙醇胺置于90mL水中,混合均匀。
A.3.2.4 钙红指示剂(1%):称取1g 钙红指示剂(C21O7N2SH14),加99g固体氯化钠,于研钵中混匀研细,盛于棕色广口瓶中,备用。
A.3.2.5 乙二胺四乙酸二钠标准滴定溶液(0.01mol/L):参照GB/T601‐2002中4.15配制
A.3.3 仪器和设备
A.3.3.1 碱式滴定管(50mL)。
A.3.3.2 万用电阻炉。
A.3.4 分析步骤
A.3.4.1试样消化
取测定灰分项下的坩埚连同遗留残渣,小心加入混合酸消化液25mL±5mL,置于通风橱内万用电阻炉上小火加热,酸液过少时可反复补加少量混合酸消化液继续加热消化,至消化液无色透明为止。此时消化液中可能会残留剩余酸消化液,为将其蒸出,应将消化液继续加热;若消化液较少,可反复补充10mL±5mL水缓慢加热,直至不再有高氯酸的白烟冒出,即可取下;待冷却后,将坩埚内消化液小心转移至250mL容量瓶中,并用少量水反复冲洗坩埚,并不断用PH试纸检测,直至洗涤液不再呈明显酸性为止,洗液并入容量瓶并定容。
取与消化试样相同量的混合酸消化液,按上述操作做试剂空白试验。
A.3.4.2测定
A.3.4.2.1试样及空白滴定
分别移取5mL 试样消化液及空白于250mL三角瓶中,加50mL蒸馏水混合均匀,加入2mol/L 氢氧化钾溶液5mL,再加入10%三乙醇胺1mL,加钙红指示剂0.1g,充分振摇。使溶液混合均匀,在不断振摇下,用0.01 mol/L乙二胺四乙酸二钠标准滴定溶液滴定,溶液由酒红色变为纯蓝色,即为终点。
A.3.4.2.2 结果计算
海藻酸钙含量(以氧化钙计,以干基计)的质量分数,按式(A.1)计算: ![]()
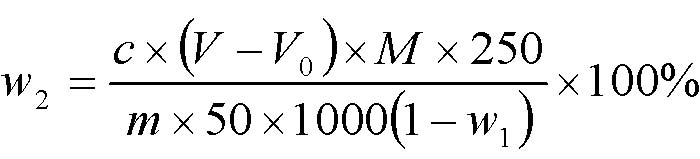 ………………………………(A.1)
………………………………(A.1)
式中:
m——试样的质量,单位为克(g);
c——乙二胺四乙酸二钠标准滴定溶液的浓度,单位为摩尔每升(mol/L);
V——滴定试样所消耗的乙二胺四乙酸二钠标准滴定溶液的体积,单位为毫升(mL);
V0——滴定空白所消耗的乙二胺四乙酸二钠标准滴定溶液的体积,单位为毫升(mL);
250——容量瓶的容积,单位为毫升(mL);
5——移取试样消化液的体积,单位为毫升(mL);
M ——氧化钙的摩尔质量,单位为克每摩尔(g/mol)[M (CaO)=56.08];
1000——换算因子;
w1—— 试样的干燥减量的质量分数,单位为百分比(%)。
试验结果以平行测定结果的算术平均值为准。在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的3.0%。
___________________________
- 二、
-
皂树皮提取物
英文名称:Quillaia extract
功能分类:乳化剂
- (一)
-
用量及使用范围
|
食品分类号 |
食品名称 |
最大使用量(g/kg) |
备注 |
|
14.02.03 |
果蔬汁(浆)类饮料 |
0.05 |
按皂素计,固体饮料按稀释倍数增加使用量 |
|
14.03 |
蛋白饮料 |
0.05 |
按皂素计,固体饮料按稀释倍数增加使用量 |
|
14.04 |
碳酸饮料 |
0.05 |
按皂素计,固体饮料按稀释倍数增加使用量 |
|
14.07 |
特殊用途饮料 |
0.05 |
按皂素计,固体饮料按稀释倍数增加使用量 |
|
14.08 |
风味饮料 |
0.05 |
按皂素计,固体饮料按稀释倍数增加使用量 |
- (二
-
) 质量规格要求
1 范围
本质量规格要求适用于以皂树(Quillajasaponaria Molina)的树皮、树干或枝条为原料,磨碎后使用水溶剂提取法提取出来经净化、精制等工艺生产的食品添加剂皂树皮提取物。商品化的皂树皮提取物产品可为液体或粉末状,粉末状产品可含有例如乳糖、麦芽糖醇、麦芽糊精、糊精、聚葡萄糖等作为载体。液体产品可以使用苯甲酸钠或乙醇以便保存。
2 产品分类
皂树皮提取物按照不同皂素含量范围分为1型和2型两种产品类型。
3 技术要求
3.1 感官要求:应符合表1的规定。
表1 感官要求
|
项 目 |
指 标 |
检验方法 |
|
状态 |
液体或粉末状 |
取适量样品置于白色瓷盘中,于自然光线下采用目测的方法观察状态、色泽及杂质,采用鼻嗅的方法闻其气味。 |
|
色泽 |
浅棕色至棕色 |
|
|
气味 |
具有皂树皮提取物特有的气味 |
|
|
杂质 |
无肉眼可见外来杂质 |
3.2 理化指标:应符合表2的规定。
表2 理化指标
|
项目 |
指标 |
检测方法 |
|
|
1型 |
2型 |
||
|
水分,w/% ≤ (限粉末形态产品) |
6 |
GB 5009.3 卡尔•费休法 |
|
|
干燥失重,w/% (限液体形态产品) |
50~80 |
50~90 |
GB 5009.3直接干燥法 |
|
pH |
3.7~5.5 |
附录A中A.3 |
|
|
灰分(以干基计),w/% ≤ |
14 |
5 |
GB 5009.4a |
|
丹宁酸(以干基计),w/%≤ |
8 |
附录A中A.4 |
|
|
皂素含量(以干基计),w/% |
20~26 |
65~90 |
附录A中A.5 |
|
铅(Pb)/(mg/kg) ≤ |
2.0 |
GB 5009.12 |
|
|
a粉状样品,使用1.0g;液体样品,使用干燥失重后的残余物 |
|||
附录A
检验方法
A.1 一般规定
本质量规格要求除另有规定外,在色谱分析中均使用色谱纯试剂和GB/T 6682中规定的一级水,其余检验所用试剂的纯度均为分析纯,试验用水应符合GB/T 6682中三级水的规定。
A.2 鉴别试验
A.2.1 试样应具有较强水溶性,但不溶于乙醇、丙酮、甲醇和丁醇。
A.2.2 称取0.5g粉末试样溶解在9.5g水中或量取1mL液体试样溶解在9mL水中,加入装有350mL水的1000mL量筒中,量筒加盖后用力摇晃30次后静置,30min后记录泡沫体积(mL),泡沫体积应达到150mL。
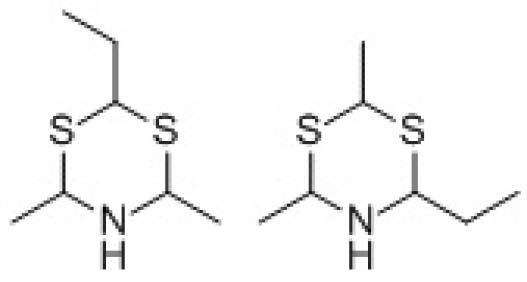
A.2.3 按照皂素含量高效液相色谱测定法(A.5),试样主峰的保留时间应与皂素标准品的皂素主要组分(QS‐18)主峰一致。
A.2.4观察A.2.2中粉末试样溶液,溶液不应出现浑浊。将该溶液置于在520nm波长下测定其吸光度,吸光度应小于1.2。(此方法仅限粉末试样)
A.3 pH的测定
A.3.1 仪器和设备
pH计。
A.3.2 操作步骤
A.3.2.1 调整pH计
按仪器使用说明书调试和校正pH计。
A.3.2.2 测定
称取适量试样,用水配制成4%(w/%)的皂树皮提取物待测液。然后用水冲洗电极探头,用滤纸轻轻吸干,将电极插入待测溶液中,调节温度调节器,使仪器指示温度与溶液温度相同,稳定后读数。
A.4 丹宁酸的测定
A.4.1 试剂和材料
A.4.1.1乙酸。
A.4.1.2聚乙烯聚吡咯烷酮。
A.4.2 仪器和设备
A.4.2.1电热恒温干燥箱。
A.4.2.2 离心机。
A.4.3 分析步骤
|
称取3.0g粉末试样,或含有3.0g固形物(使用干燥失重数值换算)的液体试样,精确至0.01 g,溶解在250mL水中,用乙酸调整pH为3.5,量取25 mL溶液,在105℃的温度条件下,干燥5 h,冷却,称重(m1)。量取50 mL溶液与360 mg聚乙烯聚吡咯烷酮混合,在室温下搅拌30 min,然后以3000 rpm的转速离心10 min。收集上层清液,并在105℃的温度条件下干燥5h,冷却,称重(m2)。 |
|
A.4.4 结果计算 丹宁酸(以干基计)的质量分数w按式(A.1)计算: |
|
……………(A.1) |
式中:
m1——加入聚乙烯聚吡咯烷酮前的溶液干燥后的质量,单位为克(g);
m2——加入聚乙烯聚吡咯烷酮后的溶液干燥后的质量,单位为克(g);
2——换算系数。
试验结果以平行测定结果的算术平均值为准。在重复性条件下获得的两次独立测定结果的绝对差值不大于5.0 %。
A.5 皂素含量的测定
A.5.1 测定方法原理:
使用高效液相色谱将皂素的主要组分QS‐7、QS‐17、QS‐18和QS‐21分离,皂树皮提取物中皂素总水平以QS‐7、QS‐17、QS‐18和QS‐21总量计算。
A.5.2 试剂和材料
A.5.2.1 皂素标准品(或类似皂素含量已知的皂素标准品)。
A.5.2.2 三氟乙酸。
A.5.2.3 高效液相色谱级乙腈。
A.5.2.4 0.2µm孔径的过滤膜。
A.5.3 仪器和设备
高效液相色谱仪:配备紫外检测器。
A.5.4 参考色谱条件
A.5.4.1 色谱柱:C4键合硅胶色谱柱(长4.6×250 mm,孔径300A, 粒径5μm)或其他等效色谱柱。
A.5.4.2 柱温:室温。
A.5.4.3 进样方式:梯度进样。
A.5.4.4 流动相:
流动相A:将0.15%三氟乙酸溶解于高效液相色谱级用水;
流动相B:将0.15%三氟乙酸溶解于高效液相色谱级乙腈。
A.5.4.5流速:1.0 mL/min。
A.5.4.6 检测波长:220nm。
A.5.4.7 梯度洗脱条件见表A.1。
表A.1 梯度洗脱条件
|
时间(min) |
流动相 A% |
流动相 B% |
流速(mL/min) |
|
0 |
70 |
30 |
1.0 |
|
40 |
55 |
45 |
1.0 |
|
45 |
70 |
30 |
1.0 |
A.5.4.8 进样体积:20 μL。
A.5.5 分析步骤
A.5.5.1 试样溶液的制备
A.5.5.1.1 粉末试样
称取0.5g试样,精确至0.001 g,在9.5g水中溶解,使用0.2µm孔径的过滤膜进行过滤,制备好的试样溶液约为10mL。
A.5.5.1.2 液体试样
称取1.0g试样,精确至0.001 g,用9mL水稀释,使用0.2µm孔径的过滤膜进行过滤,制备好的试样溶液约为10mL。
A.5.5.2 标准溶液的制备
称取1.5g皂素标准品,精确至0.001 g,在100mL水中溶解,使用0.2µm孔径的过滤膜进行过滤。
|
|
|
|
|
A.5.6 结果计算 A.5.6.1 根据上述样品制备方法制备的溶液中皂素的含量Csap,单位为毫克每毫升(mg/mL),按式(A.2)计算:
|
|
……………(A.2) |
|
|
|
式中: C标准——标准品的皂素浓度,单位为毫克每毫升(mg/mL)(例如C标准=13.5 mg/mL表示:1.5g标准样品中皂素含量为90%); A样品——试样中4个主要皂素组分QS‐7、QS‐17、QS‐18和QS‐21峰面积的总和,如附录B中示意图所示(皂素的主峰将在多酚主峰出现后出峰,参见附录B中图B.2所示)。 A标准——标准品中4个主要皂素峰面积的总和; 试验结果以平行测定结果的算术平均值为准。在重复性条件下获得的两次独立测定结果的绝对差值不大于2.0 %。 A.5.6.2 试样中皂素的质量分数w1按式(A.3)计算: |
|
……………(A.3)
|
|
式中: Csap——试样溶液中的皂素含量,单位为毫克每毫升(mg/mL); m样品——从制备的样品中取出的样品质量,单位为毫克(mg); v样品——制备的试样溶液体积,单位为毫升(mL)。 试验结果以平行测定结果的算术平均值为准。在重复性条件下获得的两次独立测定结果的绝对差值不大于2.0 %。 |
附录B
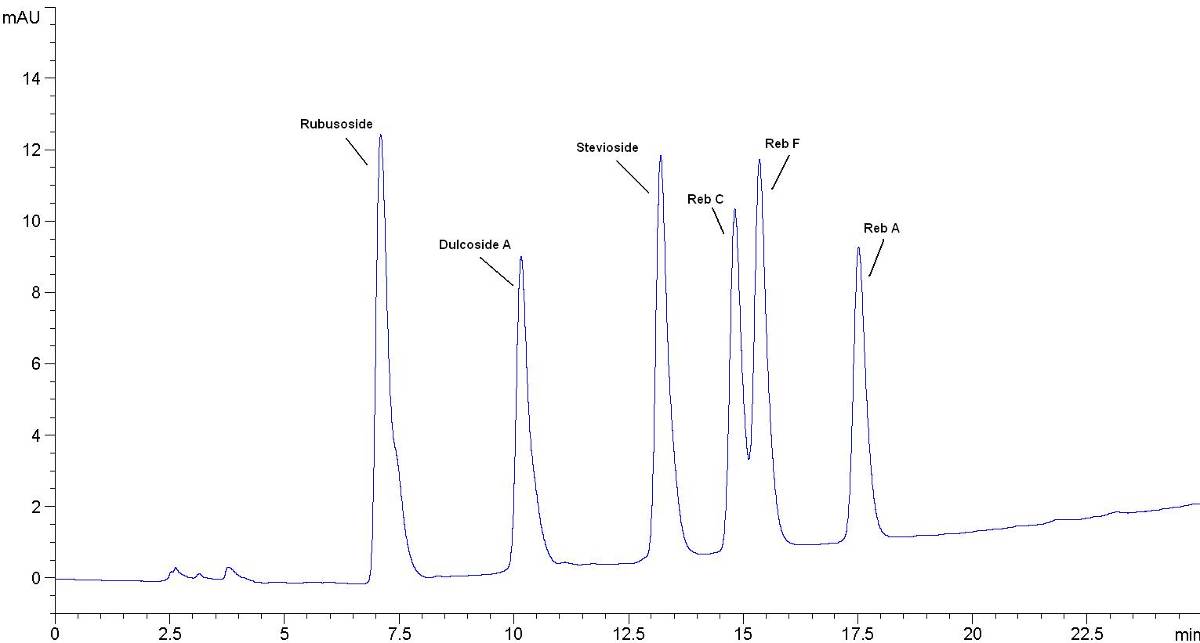
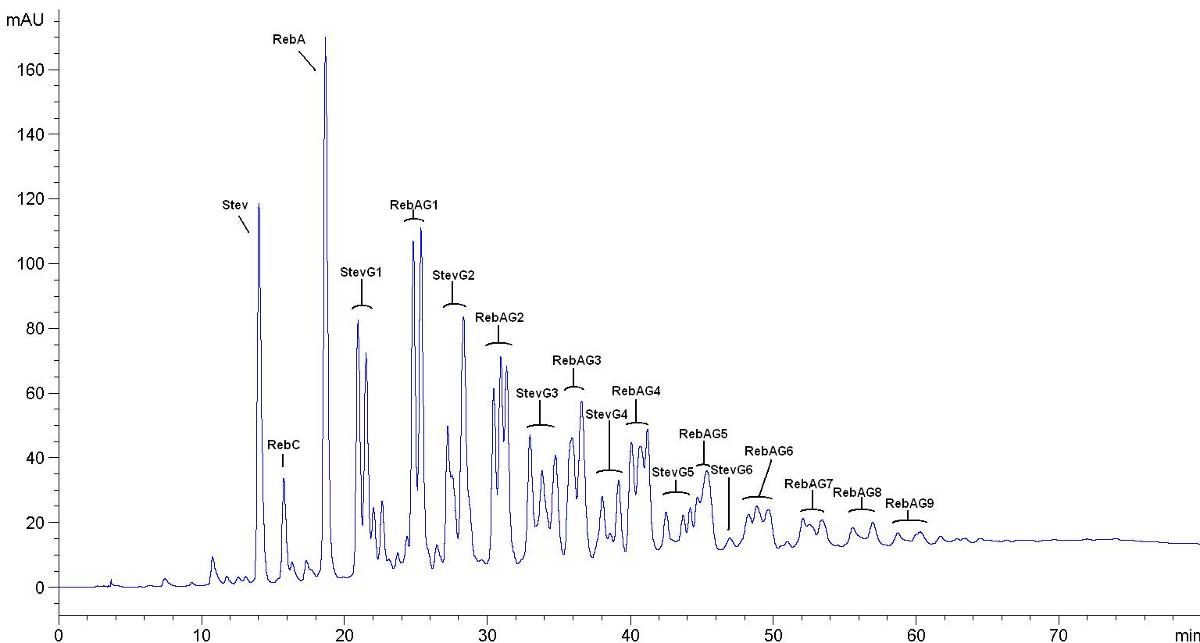
皂树皮提取物高效液相色谱示意图
B.1皂素标准品色谱图
皂素标准品色谱图见图B.1。
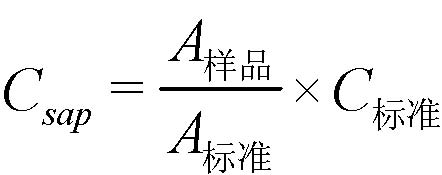
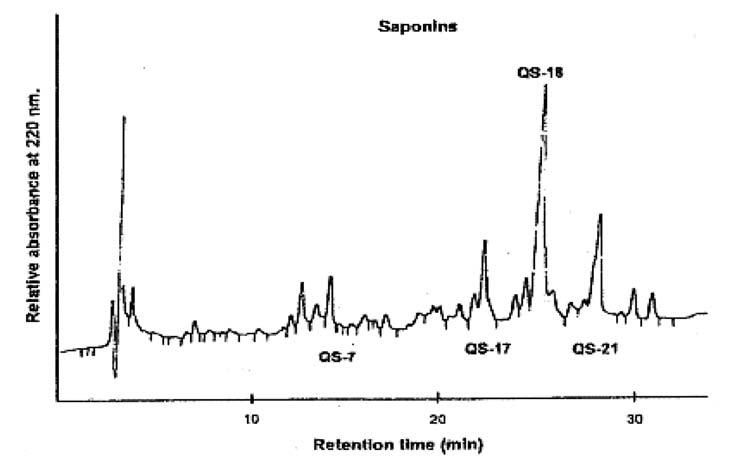
图B.1皂素标准品色谱图(15mg/mL干物质含量,相当于13.5mg/mL皂素含量)
B.2皂树皮提取物(1型)的色谱图
皂树皮提取物(1型)的色谱图见图B.2。皂树皮提取物(2型)的色谱图参照皂树皮提取物(1型)色谱图。
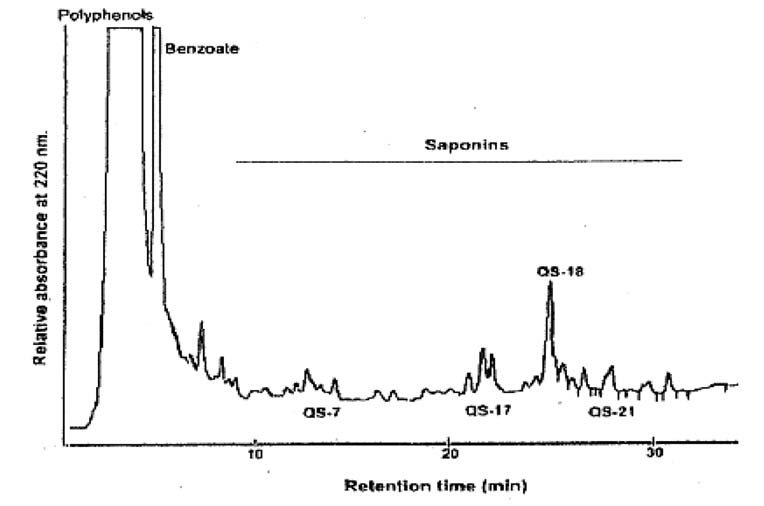
图B.2皂树皮提取物(1型)的色谱图(约55 mg/mL干物质含量)
__________________________
保留时间(min)
220纳米波长下的相对吸光度
皂素
220纳米波长下的相对吸光度
多酚
苯酸盐
皂素
保留时间(min)
- 三、
-
磷酸(湿法)
英文名称:Phosphoric acid (Wet process)
功能分类:酸度调节剂
(一)用量及使用范围
|
食品分类号 |
食品名称 |
最大使用量/(g/kg) |
备注 |
|
14.04.01 |
可乐型碳酸饮料 |
5.0 |
以PO43-计 |
(二)质量规格要求
1 范围
本质量规格要求适用于经溶剂萃取、除杂和精制制得的食品添加剂磷酸(湿法)。
2 分子式和相对分子质量
2.1 分子式
H3PO4
2.2 相对分子质量
97.99
3 技术要求
3.1 感官要求:应符合表1的规定。
表1 感官要求
|
项 目 |
要 求 |
检验方法 |
|
色泽 |
无色透明或略带浅色 |
取适量试样,置于清洁、干燥的比色管中,在自然光线下,目视观察其色泽和状态。 |
|
状态 |
稠状液体 |
3.2 理化指标:应符合表2的规定。
表2 理化指标
|
项 目 |
指 标 |
检验方法 |
|
|
磷酸(H3PO4)含量,w/% |
|
75.0~86.0 |
GB 1886.15 |
|
色度,黑曾 |
≤ |
20 |
GB/T 605 |
|
总有机碳(以C计),w/% |
≤ |
0.006 |
附录A中A.4 |
|
易氧化物(以H3PO3计),w/% |
≤ |
0.008 |
GB 1886.15 |
|
硫酸盐(以SO4计),w/% |
≤ |
0.01 |
附录A中A.5 |
|
氯化物(以Cl计),w/% |
≤ |
0.0007 |
GB/T 2091 |
|
铁(以Fe计),w/% |
≤ |
0.001 |
附录A中A.6 |
|
砷(以As计)/(mg/kg) |
≤ |
0.5 |
GB 1886.15 |
|
氟化物(以F计)/(mg/kg) |
≤ |
10 |
GB 1886.15 |
|
铅(Pb)(w)/(mg/kg) |
≤ |
2.0 |
SN/T 2049 |
|
镉(以Cd计)/(mg/kg) |
≤ |
2.0 |
SN/T 2049 |
|
汞(以Hg计),w/% |
≤ |
0.0001 |
附录A中A.7 |
|
重金属(以Pb计)/(mg/kg) |
≤ |
5.0 |
GB 1886.15 |
附录A
检验方法
A.1 安全提示
本试验方法中使用的部分试剂具有毒性、腐蚀性及易燃,操作者须小心谨慎!如溅到皮肤上应立即用水冲洗,严重者应立即治疗。使用易燃品时,严禁使用明火加热。
A.2 一般规定
本质量规格要求所用试剂和水在没有注明其他要求时,均指分析纯试剂和GB/T 6682规定的三级水。试验中所用标准溶液、杂质测定用标准溶液、制剂和制品,在没有注明其他要求时均按GB/T 601、GB/T 602、GB/T 603之规定制备。试验中所用溶液在未注明用何种溶剂配制时,均指水溶液。
A.3 鉴别试验
A.3.1 试剂和材料
A.3.1.1 氢氧化钠溶液:40 g/L。
A.3.1.2 硝酸银溶液:10 g/L。
A.3.1.3 酚酞指示液:10 g/L。
A.3.2 鉴别方法
称取约1 g试样,置于100 mL烧杯中,加10 mL水,1滴酚酞指示液,用氢氧化钠溶液调至中性,滴加硝酸银溶液,有黄色沉淀生成,该沉淀能溶于稀硝酸(5%)或氨水。
A.4 总有机碳的测定
A.4.1 方法提要
试样中的总有机碳,在过硫酸盐和紫外光的作用下,被氧化为二氧化碳,用有机碳TOC分析仪测定总碳含量。
A.4.2 试剂和材料
A.4.2.1 无二氧化碳蒸馏水或高纯水。
A.4.2.2 邻苯二甲酸氢钾标准溶液:1mL溶液含碳(C)1.0mg。称取在120℃干燥2h的基准试剂邻苯二甲酸氢钾2.1254g,加入水溶解,移入1000mL容量瓶内,用水稀释至刻度,摇匀。
A.4.2.3 高纯氮气:纯度99.999%。
A.4.3 仪器和设备
有机碳(TOC)分析仪。
A.4.4 分析步骤
A.4.4.1 工作曲线的绘制
分别移取0.00mL、1.00mL、2.00mL、3.00mL、4.00mL的邻苯二甲酸氢钾标准溶液,置于100mL的容量瓶中,用高纯水定容到刻度,摇匀。用有机碳(TOC)分析仪器制定工作曲线。
A.4.4.2 测定
称取约3g样品,精确至0.0001g。置于100mL容量瓶中,用纯水稀释至刻度,摇匀。用有机碳(TOC)分析仪测定。
A.4.5 结果计算
总有机碳(以C计)的质量分数w1按式(A.1)计算:
![]() ……………………………(A.1)
……………………………(A.1)
式中:
m1——测定试验溶液中的总有机碳的质量,单位为毫克(mg);
m—— 试样的质量,单位为克(g);
取平行测定结果的算术平均值为测定结果,平行测定结果的绝对差值不大于0.0002%。
A.5 硫酸盐的测定
A.5.1 方法提要
原子由低能级跃迁到高能级所需要的能量,是由RF发生器产生高频电磁能,通过线圈耦合到由氩气气流的矩管,从而产生等离子体。测量标准溶液所发射的特征谱线的光强,再测量待测浓度的特征谱线强度,从而确定待测溶液的浓度。
A.5.2 试剂和材料
A.5.2.1 硝酸溶液:1+1。
A.5.2.2 标准溶液:SO4标准储备液含量为1mg/mL。临用时用纯水配制成需要浓度的标准溶液。
A.5.3 仪器和设备
电感耦合等离子体发射光谱仪。
A.5.4 分析步骤
称取约3~4g试样,精确至0.0002g。置于100mL容量瓶中,加入5mL硝酸溶液,用水稀释到刻度,摇匀。在电感耦合等离子体发射光谱仪上选择SO4曲线进行测定。
A.5.5 结果计算
硫酸盐(以SO4计)的质量分数w2按式(A.2)计算:
![]() ……………………………(A.2)
……………………………(A.2)
式中:
m1——仪器读数,单位为毫克每升(mg/L);
m—— 试样的质量,单位为克(g);
取平行测定结果的算术平均值为测定结果,平行测定结果的绝对差值不大于0.0005%。
A.6 铁(以Fe计)的测定
A.6.1 试剂和材料
A.6.1.1 硝酸溶液:1+1。
A.6.1.2 标准溶液:Fe标准储备液含量为1mg/mL。临用时用纯水配制成需要浓度的标准溶液。
A.6.2 仪器和设备
电感耦合等离子体发射光谱仪。
A.6.3 分析步骤
称取约3~4g试样,精确至0.0002g。置于100mL容量瓶中,加入5mL硝酸溶液,用水稀释到刻度,摇匀。在电感耦合等离子体发射光谱仪上选择Fe曲线进行测定。
A.6.4 结果计算
Fe(以Fe计)的质量分数w3按式(A.3)计算:
![]() ……………………………(A.3)
……………………………(A.3)
式中:
m1——仪器读数,单位为毫克每升(mg/L);
m—— 试样的质量,单位为克(g);
取平行测定结果的算术平均值为测定结果,平行测定结果的绝对差值不大于0.0002%。
A.7 汞(以Hg计)的测定
A.7.1 方法提要
在酸性介质中,试样中汞被硼氢化钾(KBH4)还原成原子态汞,由载气(氩气)带入原子化器中,在特制汞空心阴极灯照射下,基态汞原子被激发至高能态,在去活化回到基态时,发射出特征波长的荧光,其荧光强度与汞含量成正比,与标准系列比较定量。
A.7.2 试剂和材料
A.7.2.1 盐酸溶液:1+1。
A.7.2.2 氢氧化钠溶液:5g/L。
A.7.2.3 硼氢化钾溶液: 称取5.0g硼氢化钾,溶于5g/L的氢氧化钠溶液中,并稀释至1000mL,混匀,现用现配。
A.7.2.4 汞标准溶液:1mL溶液含汞(Hg)0.010mg,即用即配。用移液管移取1mL按照HG/T 3696.2配制的汞标准溶液,置于100mL容量瓶中,用水稀释至刻度、摇匀。
A.7.2.5 汞标准溶液:1mL溶液含汞(Hg)0.1μg,即用即配。
A.7.2.6 氩气:纯度应大于99.99%。
A.7.3 仪器和设备
双道原子荧光光度计
A.7.4 分析步骤
A.7.4.1 仪器工作参数见表A.1。
表A.1 仪器工作参数表
|
待测 元素 |
负高压 (V) |
灯电流 (mA) |
柱高 (mm) |
载气(mL/min) |
屏蔽气 (mL/min) |
|
Hg |
230 |
15 |
10 |
300 |
900 |
A.7.4.2 标准系列溶液的配制
移取0.00mL,1.00mL,2.00mL,4.00mL,8.00mL,10.00mL Hg标准溶液(100μg /mL)于6个100mL的容量瓶中,分别加入10mL(1+1)HCl,用水稀释刻度,摇匀。在上述仪器工作参数条件下测标液的光强度,绘制标准曲线。
A.7.4.3 样品的处理和测定
称取0.5g试样(精确至0.0001g)于100mL容量瓶中,加入10mL(1+1)HCl,用水稀释至刻度,摇匀。在绘制好的工作曲线上测定。
A.7.5 结果计算
汞(以Hg计)的质量分数w4按式(A.4)计算:
![]() ……………………………(A.4)
……………………………(A.4)
式中:
c——仪器读数,单位为毫克每升(mg/L);
m——试样的质量,单位为克(g);
- 四、
-
酒石酸铁
英文名称:Iron tartrate
功能分类:抗结剂
(一)用量及使用范围
|
食品分类号 |
食品名称 |
最大使用量/(g/kg) |
备注 |
|
12.01 |
盐及代盐制品 |
0.106 |
最大使用量以酒石酸铁含量计 |
(二)质量规格要求
1 范围
本质量规格要求适用于以L-酒石酸、氢氧化钠与氯化铁为原料,经络合制得的食品添加剂酒石酸铁。
2 分子式、结构式和相对分子量
2.1 分子式
Fe (OH)2C4H4O6Na
2.2 结构式
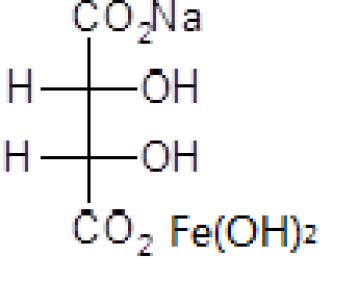
2.3相对分子量
261.93(按2007 年国际相对原子质量)
3 技术要求
3.1感官要求:应符合表1 的规定。
表1 感官要求
|
项目 |
要求 |
检验方法 |
|
色泽 |
深绿色 |
取适量试样置于50mL烧杯中,用目测法观察。 |
|
状态 |
液体 |
3.2 理化指标:应符合表2 的规定。
表2 理化指标
|
项目 |
指标 |
检验方法 |
|
|
内消旋酒石酸(以干基计二钠盐),w/% |
≥ |
37 |
附录A中A.2 |
|
D-及L-酒石酸(以干基计二钠盐),w/% |
≥ |
14 |
附录A中A.2 |
|
草酸盐(以干基计草酸),w/% |
≤ |
1.5 |
附录A中A.2 |
|
铁(Fe)(以干基计),w/% |
≥ |
8 |
GB/T 5009.90 |
|
水分,w/% |
≥ |
65 |
GB 5009.3 |
|
氯(Cl)(以干基计),w/% |
≤ |
25 |
GB/T 12457 |
|
钠(Na)(以干基计),w/% |
≤ |
23 |
GB/T 5009.91 |
|
砷(As)/(mg/kg) |
≤ |
3.0 |
GB 5009.76 |
|
铅(Pb)/(mg/kg) |
≤ |
5.0 |
GB 5009.12 |
|
汞(Hg)/(mg/kg) |
≤ |
1.0 |
GB 5009.17 |
附录A
检验方法
A.1 一般规定
本质量规格要求所用试剂和水,在没有注明其他要求时,均指分析纯试剂和GB/T 6682中规定的三级水。试验中所用标准滴定溶液、杂质测定用标准溶液、制剂及制品,在没有注明其他要求时,均按GB/T 601、GB/T 602、GB/T 603的规定制备。试验中所用溶液在未注明用何种溶剂配制时,均指水溶液。
A.2 内消旋酒石酸、D-及L-酒石酸、草酸含量的测定
A.2.1 方法提要
酒石酸铁与过量的氢氧化物反应分解,经过滤形成的 Fe(OH)3。使用有机酸色谱柱为固定相,将0.01 mol/L的硫酸作为流动相,利用液相色谱法分离组分。使用示差折光检测器检测,借助外标进行计算。
A.2.2 仪器与设备
A.2.2.1高效液相色谱仪:示差折光检测器。
A.2.2.2泵。
A.2.2.3自动进样器:配备20 µL的样品回路。
A.2.2.4分离柱:不锈钢管,长度300 mm,内径7.8 mm有机色谱柱。
A.2.2.5柱温箱。
A.2.2.6数据采集与集成系。
A.2.2.7注射器式滤器,直径30 mm,精度0.45μm,色号:绿色。
A.2.3试剂和材料
A.2.3.1硫酸,浓度0.01 mol/L。
A.2.3.2一水合内消旋酒石酸,浓度> 98 % 。
A.2.3.3D-酒石酸,浓度> 99 %。
A.2.3.4L-酒石酸,浓度> 99 %。
A.2.3.5二水合草酸,浓度> 99 %。
A.2.3.6氢氧化钠溶液,浓度5 mol/L。
A.2.4 样品
试样贮存在密闭的棕色瓶中,与氧气隔离。如果样品瓶无法装满,需充入氮气将样品覆盖。避光(紫外线)并置于冰箱低温(4°C)保存。样品溶液在2周内保持稳定。
A.2.5 分析步骤
A.2.5.1 色谱条件
A.2.5.1.1分离柱色谱柱:有机酸色谱柱,内径300 ×7.8 mm;
A.2.5.1.2柱温:10℃;
A.2.5.1.3流动相:硫酸(A.2.3.1);
A.2.5.1.4流速:0.3 ml/min;
A.2.5.1.5进样体积:20 µL;
A.2.5.1.6检测器:示差折光检测器。
A.2.5.2 标准溶液的配制
A.2.5.2.1多组分标准溶液A(2份)
向50 mL烧瓶中加入50mg至60 mg的一水合内消旋酒石酸(A.2.3.2)与20mg至30mg的D-酒石酸(A.2.3.3)或L-酒石酸(A.2.3.4),精确到0.01mg。加入50ml的硫酸(A.2.3.1)溶解。确定总质量,精确到0.1mg。
配置第二份浓度不同的多组分标准溶液A,其中,一水合内消旋酒石酸(A.2.3.2)与D-酒石酸(A.2.3.3)或L-酒石酸(A.2.3.4)的含量应有轻微区别,使测试样品的内消旋酒石酸和D-或L-酒石酸的含量在以上两个标准溶液A之间。
A.2.5.2.2草酸标准溶液B
称取250 mg的二水合草酸(A.2.3.5),精确到0.1mg,用硫酸(A.2.3.1)溶解并定容至500 mL。确定总质量,精确到1 mg。
A.2.5.3多组分标准溶液A中内消旋酒石酸、D-及L-酒石酸的浓度
按照A.2.5.6分析两个多组分标准溶液A。
用移液管抽取标准溶液A注入小玻璃瓶中,待分析。
多组分标准溶液A中内消旋酒石酸、D-及L-酒石酸的浓度分别按照公式(A.1)、(A.2)、(A.3)、(A.4)计算。
内消旋酒石酸的质量M1:
![]() …………………
…………………![]()
式中:
|
M1 |
——多组分标准溶液A中内消旋酒石酸的质量,单位为毫克(mg); |
|
Mcq1 |
——多组分标准溶液A一水内消旋酒石酸的质量,单位为毫克(mg); |
|
X |
——标准物质中内消旋酒石酸的质量百分数; |
|
150.1 |
——内消旋酒石酸的分子量; |
|
168.1 |
——一水合内消旋酒石酸的分子量; |
|
100 |
——换算因子。 |
D-及L-酒石酸的质量M2:
![]() …………………
…………………![]()
式中:
|
M2 |
——多组分标准溶液A中无水D-及L-酒石酸的质量,单位为毫克(mg); |
|
Mcq1 |
——多组分标准溶液A中一水内消旋酒石酸的质量,单位为毫克(mg); |
|
Mcq2 |
——多组分标准溶液A中无水D-或L-酒石酸的质量,单位为毫克(mg); |
|
Y |
——标准物质中无水D-及L-酒石酸的质量百分数; |
|
150.1 |
——内消旋酒石酸的分子量; |
|
168.1 |
——一水合内消旋酒石酸的分子量; |
|
100 |
——换算因子。 |
多组分标准溶液A中,内消旋酒石酸的浓度x1、D-及L-酒石酸的浓度x2, 按式(A.3)、(A.4)计算:
![]() …………………(A.3)
…………………(A.3)
![]() …………………(A.4)
…………………(A.4)
式中:
|
Mt |
——多组分标准溶液A的质量,单位为克(g); |
|
M1 |
——多组分标准溶液A中,内消旋酒石酸的质量,单位为毫克(mg); |
|
M2 |
——多组分标准溶液A中,D-及L-酒石酸的质量,单位为毫克(mg)。 |
A.2.5.4草酸标准溶液B中草酸的浓度
按照表A.1制备校准溶液(I-VII):用移液枪分别转移以下体积的草酸标准溶液B至7个50mL的烧瓶中,待分析。
表A.1 校准溶液
|
溶液(mL) |
I |
II |
III |
IV |
V |
VI |
VII |
|
草酸标准溶液B(A.2.5.2.2 ) |
0 |
0.2 |
1.0 |
2.5 |
5.0 |
7.5 |
10.0 |
添加50mL的硫酸(A.2.3.1)确定总质量,将结果精确到0.1mg。用表A.1中7个标准溶液来绘制曲线计算方程(A.2.6.1.2)。
草酸标准溶液B中草酸的浓度按公式(A.5)、(A.6)计算:
草酸的质量M3:
![]() …………………(A.5)
…………………(A.5)
式中:
|
M3 |
——草酸标准溶液B中草酸的含量,单位为毫克(mg); |
|
Mcq3 |
——草酸标准溶液B中二水合草酸的含量,单位为毫克(mg); |
|
90.0 |
——草酸的分子量; |
|
126.1 |
——二水合草酸的分子量。 |
草酸标准溶液B中,草酸浓度x3:
![]() …………………(A.6)
…………………(A.6)
式中:
|
Mt |
——草酸标准溶液B的质量,单位为克(g); |
|
M3 |
——草酸标准溶液B中草酸的质量,单位为毫克(mg); |
|
Vc |
——表A.1中,抽取的草酸标准溶液B的质量,单位为克(g); |
|
Maq |
——表A.1中,配制好的草酸标准溶液B的质量,单位为毫克(mg)。 |
A.2.5.5 测试样品
称取500mg样品,置于50 mL烧瓶中,使用25mL水稀释,加入1mL NaOH溶液(A.2.3.6),静置至少1h使得Fe(OH)3充分沉淀。确定总质量,精确到0.1 mg。测试样品溶液经注射器式滤器过滤后,注入小玻璃瓶中,待分析。
A.2.5.6测定
分别注射20 µL的多组分标准溶液A(A.2.5.2.1),草酸标准溶液B(A.2.5.4),和过滤后的测试样品溶液(A.2.5.5)到液相色谱仪中。使用折光率检测器记录液相色谱法的结果,并确定各组分的峰值面积(=A)。
A.2.6 结果计算
A.2.6.1 标准曲线绘制
A.2.6.1.1标准曲线的测量范围见表A.2。
表A.2 标准曲线的测量范围
|
组分 |
标样浓度范围 |
mTA浓溶液测量范围 |
|
内消旋-酒石酸 |
45 mg~55mg |
9%~11% |
|
D-及L-酒石酸 |
20 mg~30mg |
4%~6% |
|
草酸 |
0.05 mg~2.5mg |
0.01%~0.5% |
按照A.2.5.6,测试两份多组分标准溶液A(A.2.5.2.1),对相应峰面积进行积分。以组分q的浓度为横坐标,组分q的峰面积为纵坐标绘制标准曲线并计算回归方程式(A.7)。
A.2.6.1.2组分q(内消旋酒石酸、D-及L-酒石酸和草酸)校准函数
组分q标准曲线的截距aq和斜率bq,按照校准函数(A.7)计算:
![]() ………………… (A.7)
………………… (A.7)
式中:
|
aq |
——组分q标准曲线的截距; |
|
bq |
——组分q标准曲线的斜率; |
|
Y |
——标准样中组分q的峰面积(Ac); |
|
x |
——标准样中组分q的浓度,单位为毫克每克(mg/g),由式(A.3)(A.4)(A.6)计算。 |
A.2.6.2 测试样品中各组分q的浓度
测试样品中各组分浓度c(q)按式(A.8)计算:
![]() …………………(A.8)
…………………(A.8)
式中:
|
aq |
——组分q标准曲线的截距; |
|
bq |
——组分q标准曲线的斜率; |
|
Asq |
——测试样品溶液中组分q的峰面积; |
|
Ms |
——测试部分的质量,单位为毫克(mg); |
|
M |
——50mL烧瓶(A.2.5.5)中组分的质量,单位为克(g)。 |
A.2.7 精密度
取两次平行测定结果的算术平均值为报告结果。两次平行测定结果的绝对差值不大于0.2%。
_________________________
- 五、
-
茶黄素
英文名称:Theaflavins
功能分类:抗氧化剂
- (一)
-
用量及使用范围
|
食品分类号 |
食品名称 |
最大使用量/(g/kg) |
备注 |
|
02.0 |
脂肪,油和乳化脂肪制品 |
0.4 |
|
|
02.01 |
基本不含水的脂肪和油 |
0.4 |
|
|
04.05.02.01 |
熟制坚果与籽类(仅限油炸坚果与籽类) |
0.2 |
|
|
04.05.02.03 |
坚果与籽类罐头 |
0.2 |
|
|
05.02.01 |
胶基糖果 |
0.4 |
|
|
06.03.02.05 |
油炸面制品 |
0.2 |
|
|
06.06 |
即食谷物,包括碾轧燕麦(片) |
0.2 |
|
|
06.07 |
方便米面制品 |
0.2 |
|
|
07.0 |
焙烤食品 |
0.4 |
|
|
08.02 |
预制肉制品 |
0.3 |
|
|
08.03 |
熟肉制品 |
0.3 |
|
|
09.0 |
水产及其制品(包括鱼类、甲壳类、贝类、软体类、棘皮类等水产及其加工制品等) |
0.3 |
|
|
09.03 |
预制水产品(半成品) |
0.3 |
|
|
12.10 |
复合调味料 |
0.1 |
|
|
14.03.02 |
植物蛋白饮料 |
0.1 |
|
|
14.04 |
碳酸饮料 |
0.2 |
|
|
14.06 |
固体饮料 |
0.8 |
|
|
14.07 |
特殊用途饮料 |
0.2 |
|
|
14.08 |
风味饮料 |
0.2 |
|
|
14.09 |
其他类饮料 |
0.2 |
|
|
16.01 |
果冻 |
0.2 |
如用于果冻粉,按冲调倍数增加使用量 |
|
16.02.02 |
茶制品(包括调味茶和代用茶) |
0.2 |
|
|
16.06 |
膨化食品 |
0.2 |
|
- (二
-
) 质量规格要求
1 范围
本质量规格要求适用于以新鲜茶叶或茶多酚为原料,利用新鲜茶叶中天然含有的多酚氧化酶系,经生物发酵,乙酸乙酯浸提、食品工业用吸附树脂纯化,再经浓缩、干燥制得的食品添加剂茶黄素。
2 技术要求
2.1感官要求:应符合表1的规定。
表1 感官要求
|
项目 |
要求 |
检验方法 |
|
色泽 |
茶褐色或棕黄色 |
取适量试样置于清洁、干燥的白瓷盘中,在自然光线下,观察其色泽和状态。 |
|
状态 |
粉末 |
2.2理化指标:应符合表2的规定。
表2 理化指标
|
项 目 |
指标 |
检验方法 |
|
茶黄素,w/% ≥ |
20.0 |
附录A中A.3 |
|
咖啡碱,w/% ≤ |
5.0 |
GB/T 8312 |
|
水分,w/% ≤ |
6.0 |
GB/T 8304a |
|
总灰分,w/% ≤ |
2.0 |
GB/T 8306 |
|
砷(以As计)/(mg/kg) ≤ |
2.0 |
GB 5009.11 |
|
重金属(以Pb计)/(mg/kg) ≤ |
10 |
GB5009.74 |
|
a干燥温度和时间分别为105℃±2℃和4h。 |
||
2.3微生物指标:应符合表3的规定。
表3 微生物指标
|
项 目 |
限量(若非指定,均以/25g表示) |
检验方法 |
|
菌落总数/(CFU/g) ≤ |
1000 |
GB 4789.2 |
|
霉菌和酵母/(CFU /g) ≤ |
100 |
GB 4789.15 |
|
大肠菌群/(MPN/g) ≤ |
3.0 |
GB 4789.3 |
|
大肠埃希氏菌 |
不得检出 |
GB 4789.38 |
|
沙门氏菌 |
不得检出 |
GB 4789.4 |
附录A
检验方法
A.1 一般规定
本质量规格要求除另有规定外,所用试剂均为分析纯,所用标准滴定溶液、杂质测定用标准溶液、制剂及制品,应按GB/T 601、GB/T 602、GB/T 603的规定制备,试验用水应符合GB/T 6682的规定。试验中所用溶液在未注明用何种溶液配制时,均指水溶液。
A.2 鉴别试验
A.2.1 颜色反应
铝盐与茶黄素复合产生红色,于波长525nm具有最大吸收。
取1mL浓度为0.2mg/mL的茶黄素甲醇溶液,置于10mL容量瓶,加入2mL浓度为0.1mol/L的三氯化铝溶液,甲醇定容,充分显色20min,于525nm具有最大吸收。
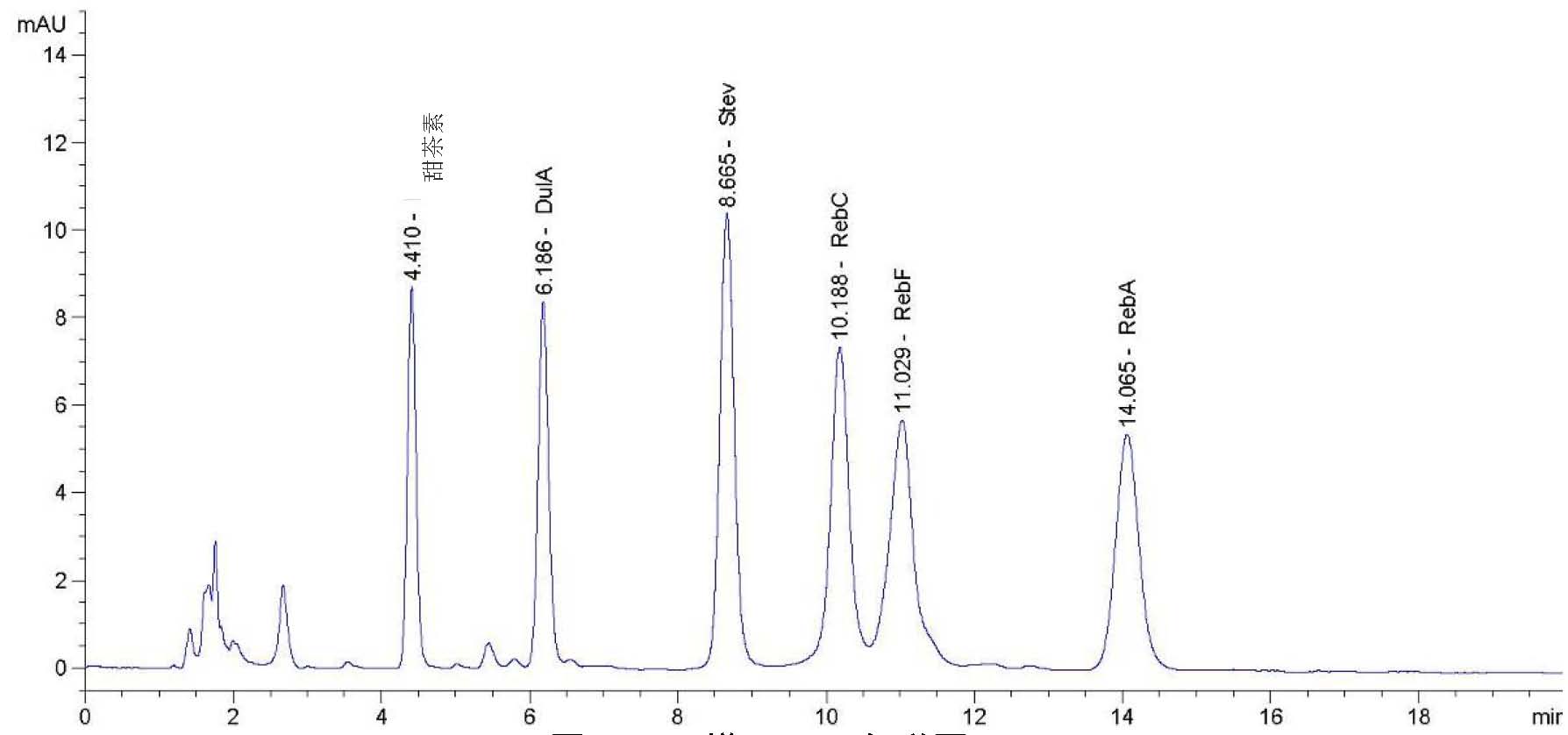
A.2.2指纹图谱分析
A.2.2.1 标准溶液的制备:称取茶黄素标准品(茶黄素含量≥80%)10mg,用95%的乙醇溶液溶解成50mL,经0.45μm的滤膜过滤。
A.2.2.2样品的制备:称取样品0.1g,用15mL乙醇溶解后移入100mL容量瓶内,用蒸馏水定容至100mL,混匀,经0.45μm的滤膜过滤。
A.2.2.3 色谱条件:
- a)
-
色谱柱:C18反相色谱柱5.0um×4.6mm×200mm;
- b)
-
流动相:A 0.1%磷酸溶液,经0.45μm的滤膜过滤;
- c)
-
柱温:35.0℃;
- d)
-
流速:2.0mL/min;
- e)
-
波长:380nm;
- f)
-
洗脱梯度见表A.1。
B乙腈(色谱纯)。
表A.1 洗脱梯度
|
时间(min) |
A% |
B% |
|
0 |
90 |
10 |
|
0.5 |
90 |
10 |
|
5 |
79 |
21 |
|
25 |
74 |
26 |
|
28 |
90 |
10 |
A.2.2.4 测定:取标准溶液和样品溶液10μL,注入色谱仪,测定,绘制标准图谱,和样品的图谱相比较。
A.2.2.5 指纹图谱见图A.1。
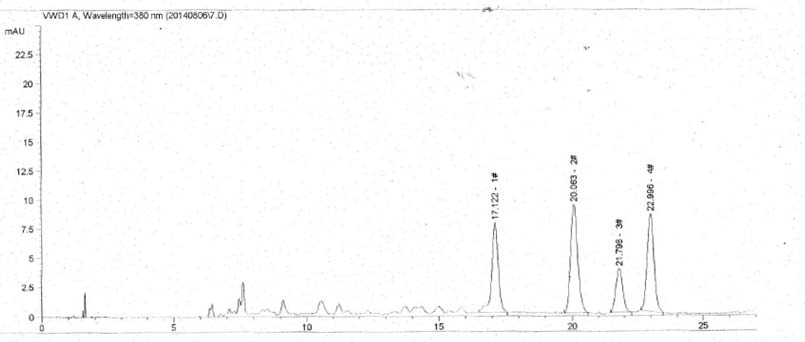
图A.1 指纹图谱
A.3 茶黄素含量的测定
A.3.1试剂和材料
A.3.1.1 95%的乙醇。
A.3.1.2 乙酸乙酯(分析纯)。
A.3.1.3 碳酸氢钠(分析纯)。
A.3.2 仪器和设备
紫外可见分光光度计。
A.3.3操作步骤
准确称量0.1g样品,用水定容至100mL,摇匀,准确移取均匀试液30mL于60mL筒形分液漏斗中,迅速加入30mL乙酸乙酯,震荡5min,静置分层,移取酯相15mL至另一30mL筒形分液漏斗中,并加入15mL现配的2.5%碳酸氢钠溶液,再震荡30s,最后移取酯相4mL至25mL容量瓶中,并加入乙醇溶液定容,充分摇匀。以乙醇溶液为空白,1cm比色杯380nm下测定吸光值A。
A.3.4 标准曲线的制作
称取80%的标准品0.1g,于100mL容量瓶中做母液。分别移取0 mL、5mL、10mL、15mL、20mL用乙醇定容至100mL,配成标准溶液,于380nm处测定吸光值,绘成标准曲线,其中斜率为a,截距为b。
A.3.5 计算
茶黄素的质量分数w按式(A.1)计算:
![]() ∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙(A.1)
∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙(A.1)
式中:
E——根据标准曲线计算的样品浓度,单位为毫克每毫升(mg/mL),E=aA+b(A为吸光值);
m——试样的质量,单位为克(g);
w1——试样的干燥失重,%;
100——定容至100mL;
25/4 ——4mL稀释到25mL;
1000 ——单位值换算1g=1000mg。
__________________________